Die Zahl klinischer Studien zu Algorithmus-basierten Innovationen ist in den vergangenen Jahren kräftig gestiegen. Angesichts des großen Potenzials von künstlicher Intelligenz für die klinische Versorgung fordern Experten regulatorische Vorgaben für die Zulassung und Langzeitbeobachtung von Algorithmus-basierten Medizinprodukten.
Von Prof. Dr. Sabine Bohnet-Joschko und Dr. Claus Zippel
Die zunehmende Entwicklung von Medizinapplikationen, die auf künstlicher Intelligenz (KI) basieren, verspricht enorme Potenziale für die Prävention, Diagnostik und Therapie von Krankheiten. Nachdem KI-basierte Gesundheitsinnovationen bislang vor allem in akademischen Einrichtungen entwickelt und erprobt wurden, streben diese nun immer stärker auf den Gesundheitsmarkt. Zu diesem Ergebnis kommt eine Arbeit der Universität Witten/Herdecke, die den aktuellen Stand bei der Registrierung und Initiierung von klinischen Studien zu Algorithmus-basierten Innovationen – die eine zentrale Voraussetzung für die Marktzulassung der Produkte darstellen – anhand der weltweit größten Datenbank für klinische Studien erforscht hat.
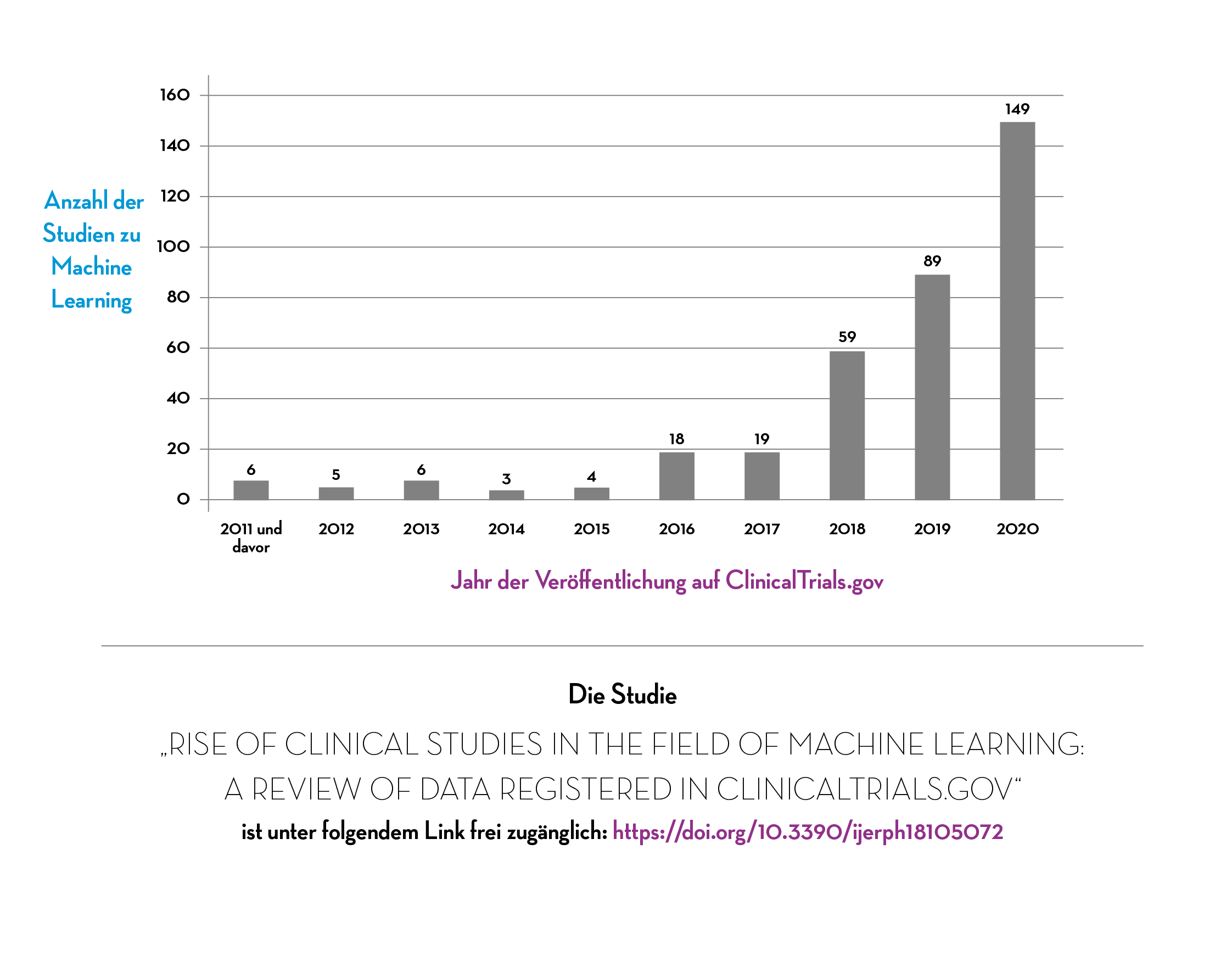
Dabei zeigte sich, dass die Anzahl der registrierten Studien zu algorithmischen Ansätzen in den vergangenen Jahren weltweit kontinuierlich gestiegen ist, mit einem besonders deutlichen Anstieg 2019 und 2020. So wurden im Jahr 2016 erst fünf Studien zu den Anwendungen registriert, während es 2019 schon 89 und 2020 bereits 149 Studieneinträge waren.
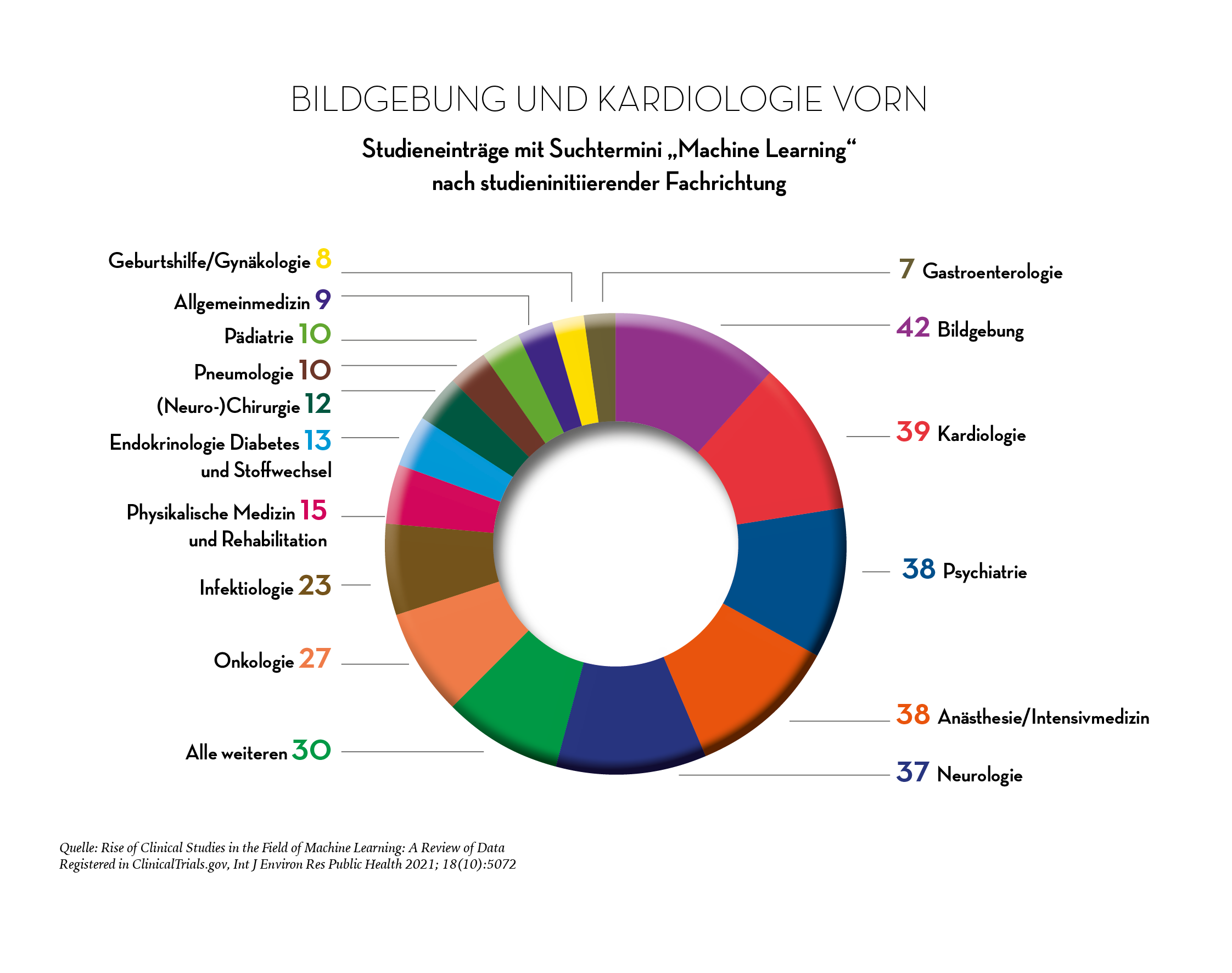
Neben dem starken Anstieg der registrierten Studien wurde auch deutlich, dass die klinisch getesteten KI-Ansätze nicht mehr nur vor allem für die assistierte Befundung von Bilddaten, sondern für ein breites Produkt- und Anwendungsspektrum und eine Vielzahl klinischer Fragestellungen entwickelt werden. Die Algorithmus-basierten Beispielanwendungen reichen von der Detektion eines Herzstillstands mit direkter Information der Rettungsleitstelle über KI-basierte Mustererkennungssysteme für die Therapiewahl in der Präzisionsonkologie bis zu Risikobewertungs- und Interventionsplattformen für psychiatrische Erkrankungen. Die meisten Studien (42) waren in der Bildgebung angesiedelt, allerdings dicht gefolgt von Kardiologie (39), Psychiatrie, Anästhesie/Intensivmedizin (je 38) und Neurologie (37). Zu einem der am stärksten gewachsenen Studienbereiche zählte die Infektiologie (23): Hier ließen sich die Studieneinträge überwiegend dem Einsatzpotenzial von KI in Forschung, Diagnostik und Therapie von Covid-19-Erkrankungen zuordnen. Insgesamt wurde auch deutlich, dass zum KI-Training neben medizinischen Bilddaten wie CT-Aufnahmen mittlerweile nahezu alle in der klinischen Versorgung verwendeten Datentypen und -sätze wie Sensordaten/EKG-Signale, Video-, Text- oder Audiodaten wie Monitorsignale herangezogen werden.
Beides, sowohl das breite Produkt- und Anwendungsspektrum als auch die zunehmende Spezialisierung Algorithmus-basierter Anwendungen, stellt die Zulassungsbehörden (und auch besonders kleine und mittelgroße Hersteller) vor enorme Herausforderungen. Die KI-Anwendungen adressieren oftmals unmittelbar eine Software oder werden in Verbindung mit einem medizinischen Gerät genutzt. Daher gelten sie aus regulatorischer Sicht zumeist als Medizinprodukt und unterliegen den Vorgaben der zur Stärkung der Patientensicherheit seit Mai 2021 in Kraft getretenen EU-Medizinprodukteverordnung (MDR).
Die EU-Verordnung benennt zwar zur klinischen Versorgung eingesetzte Software explizit als Medizinprodukt, lässt jedoch die detaillierte Ausgestaltung der regulatorischen Rahmenbedingungen für die spezifische Zulassung von KI-Anwendungen in der Gesundheitsversorgung vergleichsweise offen. So gibt es bislang kaum detaillierte Vorgaben oder harmonisierte Standards für die Bewertung, Zulassung und Langzeitbeobachtung von KI-Gesundheitssoftware. Ganz besonders gilt dies für solche Anwendungen, die auf fortlaufend lernenden Algorithmen basieren und sich nach der Zulassung bei ändernden Datensätzen im Zeitablauf anpassen können. Verkompliziert wird dies dadurch, dass die Systeme teils regelbasiert und teils lernbasiert (also selbstsuchend nach Lösungen) programmiert werden. Bei Letzterem stehen neben regulatorischen Aspekten auch viele ethische Fragen im Mittelpunkt.
Vor diesem Hintergrund ist von großem Interesse, dass die US-amerikanische Food and Drug Administration (FDA) kürzlich einen „Artificial Intelligence/Machine Learning-Based Software as a Medical Device (SaMD)“-Aktionsplan veröffentlicht hat. Er soll unter anderem schrittweise zu einem Regelungsrahmen für die klinische Prüfung und Zulassung von Medizinprodukten mit auf Basis veränderter Datensätze weiterlernenden Algorithmen führen. Als ein Hauptziel nennt der Aktionsplan, spezifische Methoden zur Stärkung der Robustheit und Vermeidung von Verzerrungen in Trainingsdatensätzen (Bias) zu entwickeln. Dadurch sollen unter anderem diskriminierende Entscheidungen von Softwareanwendungen in Bezug auf Geschlecht, soziale Herkunft und weitere Aspekte vermieden werden. Ein zweiter Schwerpunkt adressiert Aspekte von Informationssicherheit und Datenschutz, die durch eine „Gute Praxis Maschinellen Lernens“ (Good Machine Learning Practice, GMLP) gestärkt werden sollen. Ein dritter Schwerpunkt zielt darauf, Algorithmen-spezifische Parameter zu identifizieren, die in der Praxis durch Real-Word-Daten zeitnah erhoben und validiert werden sollen. Dies ist unter anderem wichtig, weil sich die klinische Entwicklung und Etablierung KI-basierter Medizinsoftware durch die voranschreitende Gewinnung qualitativ hochwertiger Patientendatenmengen zunehmend beschleunigt. Die US-Behörde entwickelt den Aktionsplan im Dialog mit diversen Interessengruppen wie Patientenvertretern und Anwendern. Dadurch soll die Akzeptanz der in der Praxis oftmals auf Skepsis treffenden KI-Anwendungen im Markt erhöht werden. Hierbei spielen etwa Fragen zur informationellen Selbstbestimmung sowie zum Daten-besitz und -schutz eine zentrale Rolle.
Für die Behörden in Europa im Allgemeinen und in Deutschland im Besonderen bieten diese Entwicklungen interessante Ansatzpunkte. Auf EU-Ebene könnte dies etwa als Impuls zur Medizinprodukt-spezifischen Konkretisierung der EU-MDR wie auch eines jüngst im April von der EU-Kommission vorgelegten Koordinationsplans mit Grundzügen zum Einsatz von KI in der EU insgesamt dienen. National bieten sich aktuell konkrete Anschlussmöglichkeiten etwa beim sogenannten offenen Fragenkatalog „Künstliche Intelligenz bei Medizinprodukten“. Der Katalog wurde von der Interessengemeinchaft der Benannten Stellen veröffentlicht und soll den Benannten Stellen, die zur klinischen Prüfung und Bewertung von Medizinprodukten autorisiert sind, wie auch den Herstellern als Orientierung dienen. Angesichts der Forschungsergebnisse zur zunehmenden Anzahl klinischer Studien im Bereich Algorithmen-basierter Anwendungen sind weitreichende Anpassungen der Regulatorik weltweit und in Europa für KI-Gesundheitssoftware und -applikationen sowie die damit verbundenen Spezifika zu erwarten.
Prof. Dr. Sabine Bohnet-Joschko ist Inhaberin des Lehrstuhls für Management und Innovation im Gesundheitswesen an der Universität Witten/Herdecke, Dr. Claus Zippel ist Professor für Betriebswirtschaftslehre und Management im Gesundheitswesen an der KH Mainz und Mitglied im wissenschaftlichen Kollegium des ATLAS Digitale Gesundheitswirtschaft.
ANZEIGE